I mitocondri come elementi centrali nella risposta infiammatoria polmonare in fibrosi cistica
Un farmaco o un trattamento ideale in fibrosi cistica (FC) dovrebbe essere una molecola che genera condizioni favorevoli per ripristinare o potenziare la residua funzionalità del canale regolatore della conduttanza transmembrana della fibrosi cistica (CFTR) mutato, interrompendo inoltre il ciclo vizioso che sostiene l’esagerata infiammazione polmonare in FC.
A questo riguardo, noi abbiamo dimostrato che modificazioni nel segnale calcio (Ca2+) mitocondriale esacerbavano le risposte infiammatorie e alteravano l’autofagia nelle vie aeree in FC1,2. Tra i difetti associati al canale CFTR, il perturbato segnale Ca2+ ha un rilevante ruolo nella fisiopatologia della malattia. Modificazioni nel segnale Ca2+ sono state osservate in diverse cellule primarie umane derivanti da pazienti affetti da FC, tra i quali cellule epiteliali bronchiali e immunologiche1,3-6 (Figura 1).
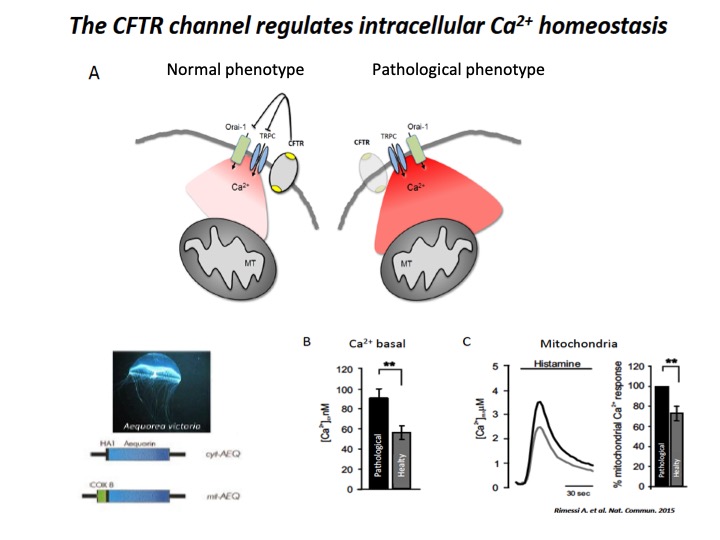
Fig. 1: Implication of CFTR in Ca2+ signaling
A) Schematic representation of intracellular Ca2+ overload in Cystic fibrosis airway cells (pathological phenotype). The cellular Ca2+ homeostasis is regulated by systems of Ca2+-entry and Ca2+-efflux located in plasma membrane and organelles, which are the principal molecular systems involved in the abnormal intracellular Ca2+ signaling associated with defective CFTR channel, where their dysfunction contribute to physiopathology of CF lung disease. B) Basal [Ca2+] ± ES in CF (pathological) vs non-CF (healthy) cells under resting conditions. C) Representative histamine-dependent mitochondrial Ca2+ response in pathological and healthy cells. The histograms show the % change of the Ca2+ responses in control compared with CF cells.
In tutte loro, la concentrazione del Ca2+ mitocondriale era aumentata rispetto a cellule non-FC, con pesanti ripercussioni sulla suscettibilità ai patogeni, i quali promuovevano disfunzione mitocondriale e indebolimento dell’autofagia, conducendo a persistenti esacerbazioni, alterazioni nelle risposte antimicrobiche ed eccessivi rilasci di mediatori pro-infiammatori nei polmoni di pazienti affetti da FC7-11 (Figura 2).
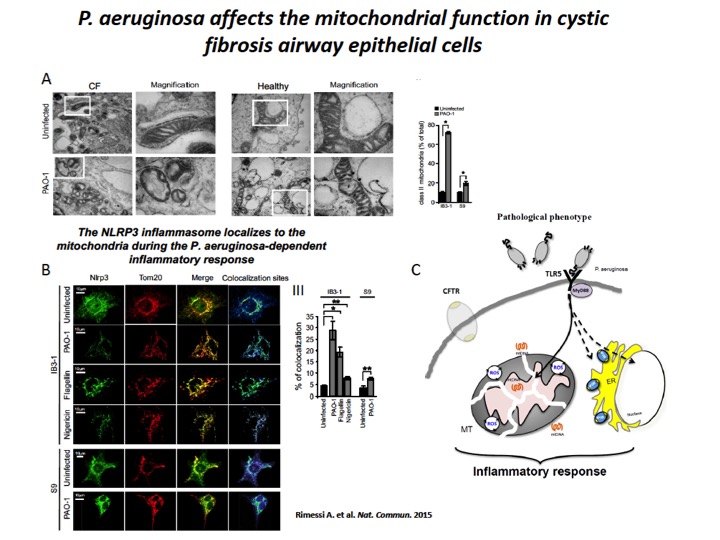
Fig. 2: P. aeruginosa induces mitochondrial dysfunction and NLRP3 activation in CF airway cells
A) The mitochondria in CF airway cells show higher susceptibility to P. aeruginosa-dependent structural perturbations than non-CF cells. Representative transmission EM fields of mitochondria of IB3-1 and S9 cells treated with the PAO-1 strain are shown (× 10,000). The white box represents × 80,000 magnification of a single mitochondrion. Morphometric analysis of randomly selected EM fields of mitochondria class II in IB3-1 and S9 cells. The data represent the % of mitochondrial class II±s.e. in reference to the total mitochondria of three independent experiments. B) P. aeruginosa induces mitochondrial NLRP3 localization and activation in CF airway cells. Confocal immunofluorescence images of IB3-1 and S9 cells endogenously expressing NLRP3. Co-localization of NLRP3 with the mitochondrial protein TOM20 during the PAO-1-, flagellin- and nigericin-dependent responses. IB3-1 cells were treated with PAO-1 (100 MOI) or flagellin (10 μg ml−1) for 2 h and with nigericin (10 μM) for 30 min. Quantification of NLRP3 co-localization with mitochondria in IB3-1 cells, expressed as the % of co-localization, which was calculated as average volume of the overlapping areas. C) Schematic representation of P. aeruginosa-triggered mitochondrial damage-associated molecular patterns (mtDAMPs) releases and consequent inflammatory activation in CF airway cells, due to mitochondrial dysfunction.
I mitocondri sono considerati centrali di segnalazione per modulare specifiche funzioni cellulari, come il metabolismo e la morte cellulare, attraverso la regolazione dell’omeostasi del segnale Ca2+ mitocondriale che normalmente avvengono mediante comunicazione inter-organello, in particolare con il reticolo endoplasmatico (ER) (Figura 3). In questi anni continue evidenze mostrano che i mitocondri agiscono come centrali regolatorie, checkpoint e arbitri per i processi infiammatori. Le disfunzioni mitocondriali e le alterazioni nel segnale Ca2+ mitocondriale sostengono l’infiammazione in diverse condizioni patologiche, inclusa la FC.
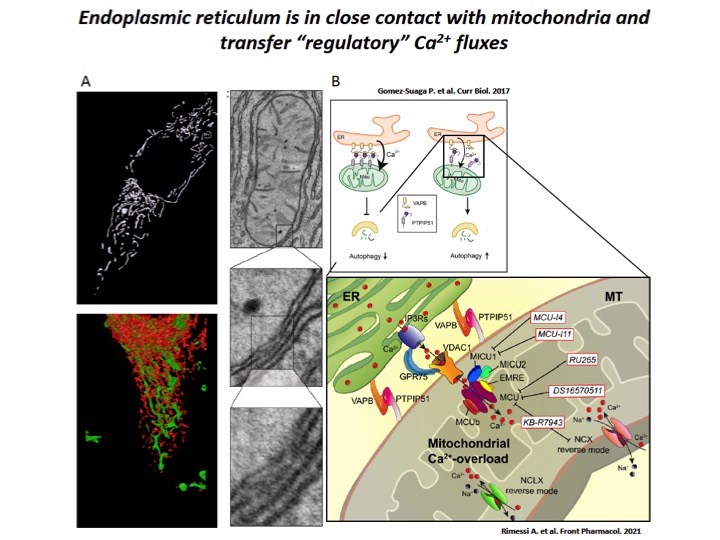
Fig. 3: Dampening the mitochondrial Ca2+-overload in cystic fibrosis
A) ER-mitochondria interfaces are intimate and dynamic regions between ER and mitochondrial outer membranes, where a series of specialized molecular bridges control the frequency of interactions, the size and the spacing between the organelles, changing at front of cellular and functional requests. High-resolution 3D imaging of mitochondria (green) and ER (red) in a HeLa cell and representative transmission EM fields of ER-mitochondria juxtaposition. B) During the recurrent pathogen infections, the increased of ER-mitochondria juxtaposition induces the dysregulation of Ca2+ signaling in CF, which causes mitochondrial Ca2+-overload in airway cells with consequent autophagic impairment and inflammation. The mitochondrial Ca2+-overload is mediated by an increased ER-mitochondria Ca2+ transfer through the IP3Rs-VDAC-MCU axis due to the stabilization of VAPB-PTPIP51 tethers. Indeed, the increased ENaC-dependent Na+ absorption due to defective CFTR in CF could stimulate NCX and NCLX exchangers to work in reverse mode triggering intracellular and mitochondrial Ca2+-influx, which may worsen the excessive mitochondrial Ca2+-uptake. To dampen the detrimental Ca2+ accumulation in matrix, a new class of mitochondrial Ca2+ modulator drugs are under investigation.
In questi anni, noi abbiamo dimostrato che durante l’infezione da P. aeruginosa in cellule polmonari di pazienti affetti da FC aumentava l’interazione tra il reticolo endoplasmatico e i mitocondri dovuto alla stabilizzazione del legame tra la proteina reticolare vesicle-associated membrane protein-associated protein B e la proteina mitocondriale tyrosine phosphatase interacting protein 51, le quali favoriscono il trasferimento del Ca2+ tra gli organelli, via Mitochondrial Calcium Uniporter (MCU)2 (Figura 4). Questo determinava disfunzione mitocondriale in cellule polmonari FC con conseguente alterazione del potenziale di membrana e produzione di radicali liberi dell’ossigeno (ROS), inducendo a persistente attivazione dell’Unfolding Protein Response mitocondriale (UPRmt) e dell’inflammasoma NLRP3. A sua volta l’attivazione di questi processi down-regolavano le risposte autofagiche selettive, quali mitofagia (degradazione autofagica selettiva di mitocondri alterati) e xenofagia (rimozione autofagica intracellulare di patogeni), favorendo la sopravvivenza dei patogeni e il peggioramento della risposta infiammatoria nei polmoni di pazienti affetti da FC 1,2 (Figura 4).
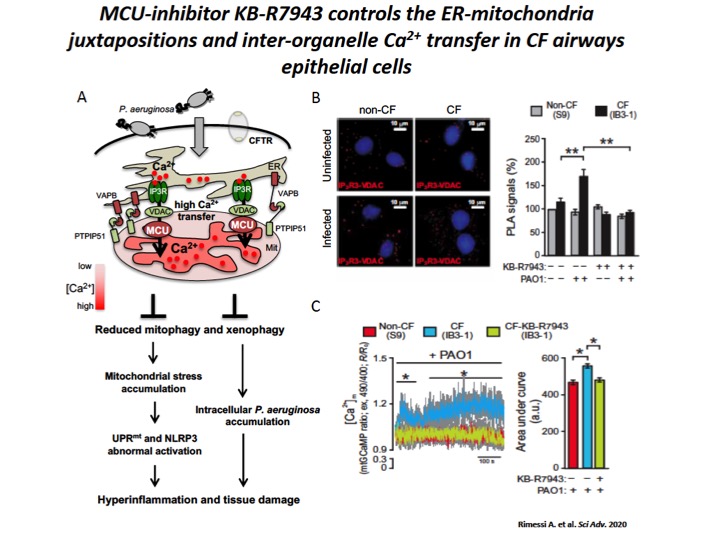
Fig. 4: The increase of ER-mitochondria tethering facilitates the inter-organelle Ca2+ transfer, via MCU, in CF airway cells during P. aeruginosa infection.
A) Schematic representation of increased inter-organelle Ca2+ transfer due by VAPB and PTPIP51 tethering stabilization in CF airway cells during P. aeruginosa infection, with relative consequences associated to selective autophagy impairments. Reduced selective autophagic responses (mitophagy and xenophagy) potentiate UPRmt and inflammasome activation, favoring a vicious cycle that sustain the hyperinflammation in CF. B) Representative images with Protein Ligation Assay (PLA) signals (red) in the different cells are shown. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole (blue). The bar chart shows quantification of IP3-R3-VDAC PLA signals (%), respect to uninfected non-CF cells. C) Mitochondrial Ca2+ dynamics in S9- (non-CF), IB3-1– (CF), and KB-R7943–treated IB3-1 (CF-KB-R7943) cells exposed to PAO1 at an MOI of 100 were evaluated through ratiometric imaging of mitochondrial-targeted GCaMP6.
Controllando il sovraccarico di Ca2+ mitocondriale mediante inibizione di MCU via KB-R7943, attenuavamo la disfunzione mitocondriale indotta da P. aeruginosa, il quale regolava l’attivazione di UPRmt e dell’inflammasoma NLRP3 con positive ricadute sulle risposte autofagiche selettive e sull’infiammazione polmonare in paziente affetti da FC sia in vitro che in vivo (Figura 5) 2. Una serie di risultati che ci hanno suggerito che il Ca2+ mitocondriale in FC può esser modulato per controllare lo sviluppo della malattia polmonare, influenzando le rispose infiammatorie e autofagiche.
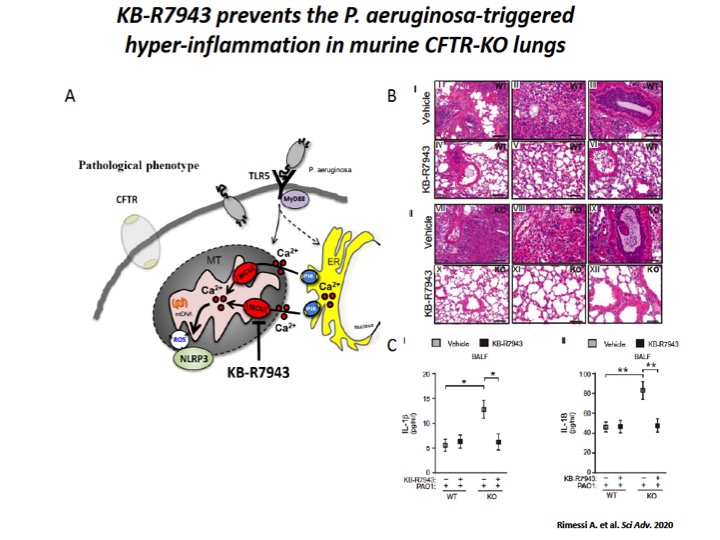
Fig. 5: The mitochondrial Ca2+ modulator, KB-R7943, limits the P. aeruginosa–triggered inflammatory response in vivo.
A) Schematic representation in which KB-R7943, inhibiting MCU, limits the activation of NLRP3 inflammasome in CF airways through the regulation of inter-organelle Ca2+ transfer via MCU. B) Reduced degree and extension of inflammation in CF lungs when pretreated with mitochondrial Ca2+ modulator, KB-R7943. The images exemplify the lungs of WT and CFTR-KO mice stained with hematoxylin and eosin (H&E), pretreated 1 hour before infection with KB-R7943 and vehicle. Scale bars, 250 μm (I, IV, VII, and X) and 500 μm (II, III, V, VI, VIII, IX, XI, and XII). C) The levels of released IL-1β (I) and IL-18 (II) measured in BALF of WT and CFTR-KO mice exposed to P. aeruginosa infection, treated with KB-R7943 and vehicle.
L’obiettivo principale del nostro progetto è acquisire evidenze precliniche di nuovi modulatori del segnale Ca2+ mitocondriale, come agenti farmacologici in grado di controllare il sovraccarico dello ione nei mitocondri e l’indebolimento dell’autofagia per prevenire l’esacerbazione dell’infiammazione polmonare in FC e cooperare con modulatori del canale CFTR per migliorare la funzionalità residua del canale stesso, sia in vitro che in vivo. Miriamo a investigare gli effetti anti-infiammatori dei nuovi modulatori del segnale Ca2+ mitocondriale, in vitro in cellule polmonari primarie di pazienti affetti FC e in vivo in modelli murini CFTR-null e DF508-CFTR.
Determineremo se i nuovi modulatori del segnale Ca2+ mitocondriale, agendo anche da modulatori autofagici, cooperano con correttori e potenziatori del canale CFTR nel migliorare la funzionalità residua dei canali mutati. Infine, indagheremo se i nuovi modulatori del segnale Ca2+ mitocondriale, salvaguardando la fisiologia dei mitocondri, influenzano l’alterata polarizzazione e la risposta antimicrobica di cellule immunologiche di pazienti affetti da FC, considerato che l’esagerata risposta infiammatoria polmonare in FC è condizionata anche dall’alterato fenotipo delle cellule immunologiche.
Ci aspettiamo che controllando il segnale Ca2+ mitocondriale contrasteremo l’amplificazione dei segnali infiammatori in cellule epiteliali respiratorie e immunologiche durante l’infezione da patogeni, preservando l’integrità dei mitocondri.
Riferimenti bibliografici
1. Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca2+ -dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun. 2015;6:6201.
2. Rimessi A, Pozzato C, Carparelli L, Rossi A, Ranucci S, De Fino I, Cigana C, Talarico A, Wieckowski MR, Ribeiro CMP, Trapella C, Rossi G, Cabrini G, Bragonzi A, Pinton P. Pharmacological modulation of mitochondrial calcium uniporter controls lung inflammation in cystic fibrosis. Sci Adv. 2020;6:eaax9093.
3. Robledo-Avila FH, Ruiz-Rosado JD, Brockman KL, Kopp BT, Amer AO, McCoy K, Bakaletz LO, Partida-Sanchez S. Dysregulated Calcium Homeostasis in Cystic Fibrosis Neutrophils Leads to Deficient Antimicrobial Responses. J Immunol. 2018;201:2016-2027.
4. Waller RL, Brattin WJ, Dearborn DG. Cytosolic free calcium concentration and intracellular calcium distribution in lymphocytes from cystic fibrosis patients. Life Sci. 1984;35:775-81.
5. Rimessi, A., Vitto, V. A. M., Patergnani, S. and Pinton, P. Update on calcium signaling in cystic fibrosis lung disease. Front in Pharmacol 2021
6. Brockman SM, Bodas M, Silverberg D, Sharma A, Vij N. Dendrimer-based selective autophagy-induction rescues ΔF508-CFTR and inhibits Pseudomonas aeruginosa infection in cystic fibrosis. PLoS One. 2017;12:e0184793.
7. Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, D’Eletto M, Tosco A, De Gregorio F, Izzo V, Maiuri MC, Kroemer G, Raia V, Maiuri L. Cysteamine re-establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis-relevant F508del-CFTR mutation. Cell Death Dis. 2017;8:e2544.
8. Pehote G, Bodas M, Brucia K, Vij N. Cigarette Smoke Exposure Inhibits Bacterial Killing via TFEB-Mediated Autophagy Impairment and Resulting Phagocytosis Defect. Mediators Inflamm. 2017;2017:3028082.
9. Shrestha CL, Assani KD, Rinehardt H, Albastroiu F, Zhang S, Shell R, Amer AO, Schlesinger LS, Kopp BT. Cysteamine-mediated clearance of antibiotic-resistant pathogens in human cystic fibrosis macrophages. PLoS One. 2017;12:e0186169.
10. Patergnani S, Vitto VAM, Pinton P, Rimessi A. Mitochondrial Stress Responses and “Mito-Inflammation” in Cystic Fibrosis. Front Pharmacol. 2020;11:581114.